
太原理工大学郭俊杰教授团队在高效酸性甲醇氧化反应(MOR)催化材料设计方面取得重要进展,于近日在材料领域顶级期刊《Advanced Functional Materials》发表了题为“Sequential Constructing Pt-Mo-NG Single-atom/Nanoparticle Cooperative Catalytic System for Boosting the Methanol Oxidation Performance”的研究论文。该论文第一署名单位为太原理工大学,第一作者是硕士研究生周贻冰,通讯作者是新材料界面科学与工程教育部重点实验室郭俊杰教授和梁浩杰讲师。
直接甲醇燃料电池(DMFCs)阳极MOR动力学缓慢,且Pt基催化剂易受CO中间体毒化。近年来,构建单原子与纳米颗粒协同催化体系为突破上述限制提供了新方向,但金属沉积顺序对二者界面相互作用及催化性能的关键调控作用长期被忽视,其内在协同机制尚未得到系统阐明。
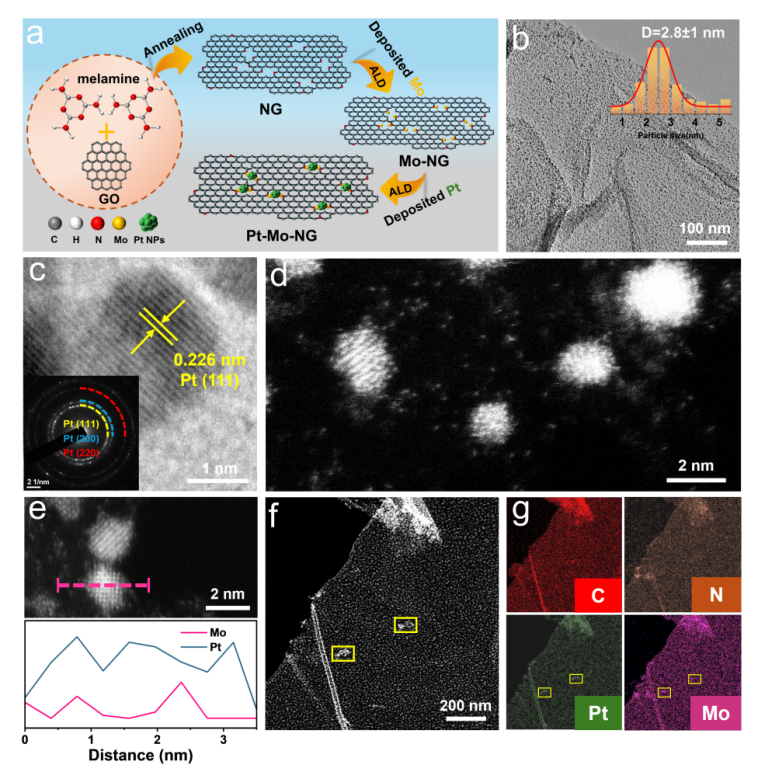
图1 Pt-Mo-NG协同催化剂的合成和显微结构表征
本工作提出了一种基于原子层沉积(ALD)技术精确控制沉积顺序,构建Mo单原子与Pt纳米粒子协同催化体系的新策略。结构表征表明,预沉积的Mo单原子可作为Pt纳米颗粒的优先成核位点,有效抑制Pt的迁移与团聚,使Pt纳米颗粒平均尺寸仅为2.80 nm,远小于逆序制备的Mo/Pt-NG(4.60 nm)和Pt-NG(4.70 nm)。
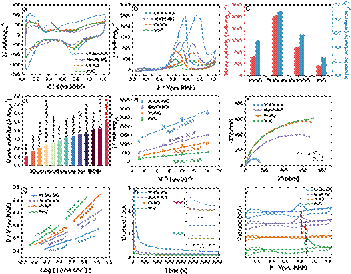
图2 Pt-Mo-NG协同催化剂的MOR性能
电化学性能测试显示,Pt-Mo-NG催化剂在酸性MOR中表现出卓越的催化活性与稳定性。其质量活性高达2037.76 mA・mgPt⁻¹,是商用20% Pt/C的5.82倍。同时,该催化剂展现出优异的抗CO中毒能力和循环稳定性,CO氧化峰电位较Pt/C负移91 mV。值得注意的是,Pt-Mo-NG在乙醇氧化反应(EOR)中同样表现出优异的催化性能。
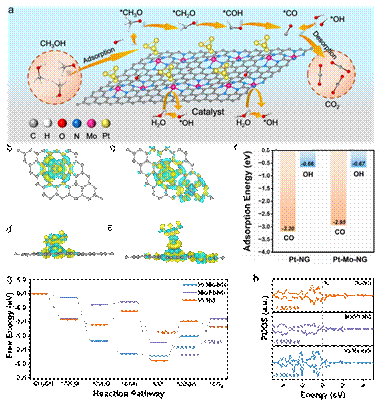
图3 Pt-Mo-NG协同催化剂的DFT计算
结合实验和DFT理论计算,研究团队深入揭示了沉积顺序调控催化性能的内在机制。预沉积的Mo单原子通过电子转移效应优化了Pt的d带中心,既保证了甲醇分子的高效脱氢活化,又显著弱化了Pt对CO中间体的吸附强度。同时,孤立的Mo单原子可促进水分子的解离生成高活性*OH物种,加速CO中间体的氧化脱除,从而提高了催化剂的抗CO中毒能力。
该工作首次揭示沉积顺序对单原子-纳米颗粒协同体系微观结构、电子效应及催化性能的关键作用,提出单原子桥连催化剂设计新策略,为构筑高效稳定低成本Pt基燃料电池阳极催化剂提供新思路。